ターナー症候群について知っておくべきすべてのこと


X染色体の完全または部分的な欠如を特徴とする、性染色体に影響を及ぼす遺伝性疾患であるターナー症候群ですが、その核型は45Xであり、女性が発症する疾患です。
単一のX染色体の存在による低身長や第二次性徴の遅れを契機にこの疾患を疑われ、染色体検査により確定診断されます。知的発達は正常であるものの、対人ストレスを強く感じる傾向があります。
統計
統計的には、ターナー症候群の症例の50%が、第2染色体の全損失がある45Xの核型を保持しています。また 15%は、この染色体の部分的な喪失であるため、この場合は染色体の長い部分または短い部分のいずれかが影響を受ける可能性があります。
残りのケースはモザイク型で、 遺伝的変化を伴う2つ以上の細胞系の存在を特徴としています。 このため、ターナー症候群を発症している人は、遺伝的に正常ないくつかの細胞(45XX)および遺伝的に変化した細胞(45X)をいくつか保有します。
正常な状態の性染色体
正常な条件下では、一般的に23組の染色体を持っています。 これらのうち22は、非性的常染色体または染色体に対応し、最後のペアは性的対となります。
性染色体は父親と母親から継承され、赤ちゃんの遺伝的性別を決定します。2つのX染色体(46XX)の存在は、性別が女性であることを決定し、これが女性の第一次そして第二次性徴を発現させます。
Y染色体(46XY)の存在は、性別が男性であることを決定するため、これが男性的な性的特性および第二次性徴の発現につながります。
ターナー症候群と診断されたら?
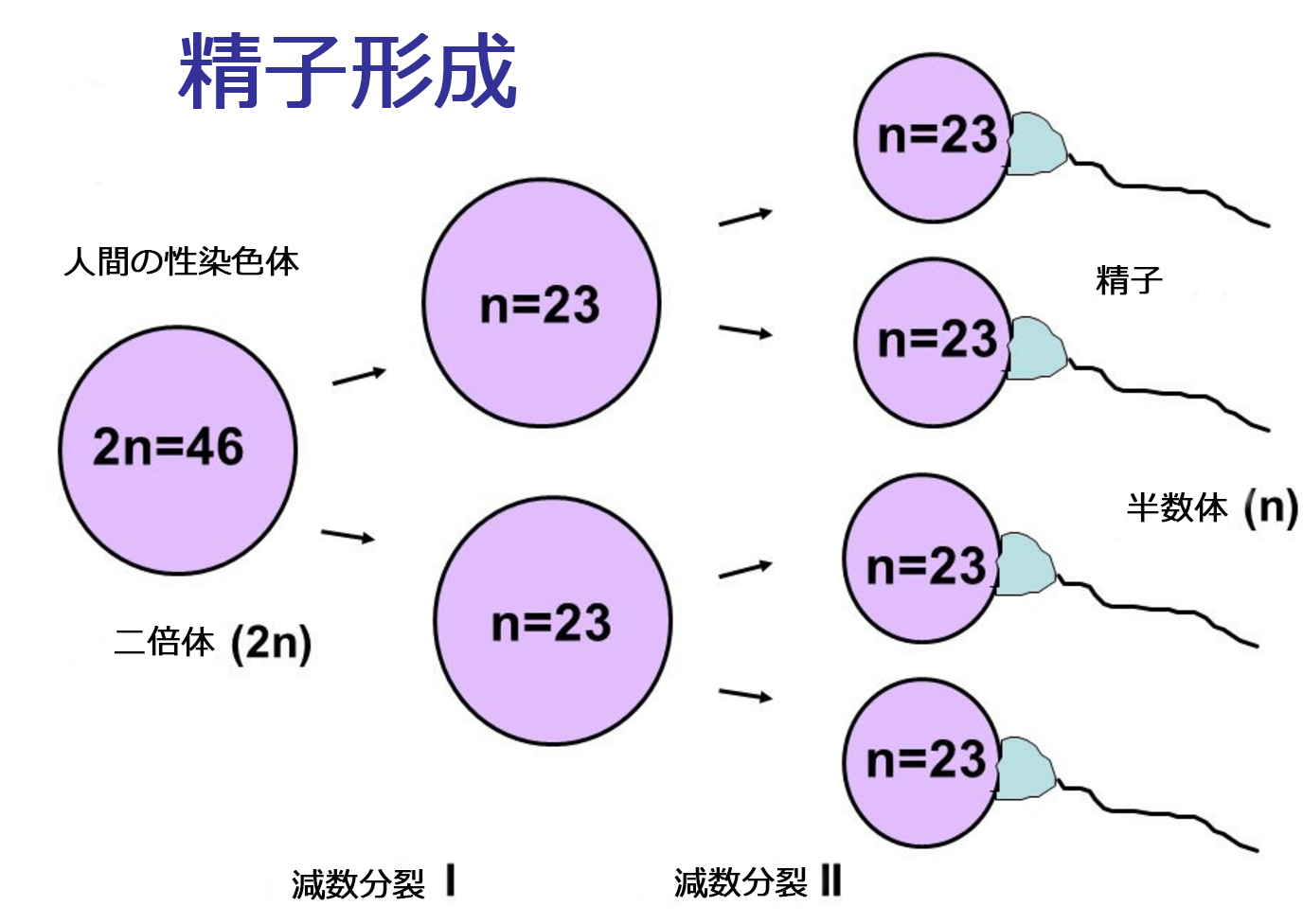
ターナー症候群と呼ばれる染色体異常の説明に入る前に、生殖細胞分裂(配偶子)の特定について少しご紹介します。
生殖細胞分裂(配偶子)の特定プロセスは減数分裂として知られており、減数分裂Iおよび減数分裂IIとして知られる2つの段階で生じます。
全体的な目標は、元の細胞からの遺伝情報の半分の娘細胞を生成することです:
- 基本的には、23組の染色体を含む幹細胞から始まる
- 減数分裂後Iには、それぞれ23の染色体を有する2つの娘細胞が存在する
- 減数分裂IIの後、これら2つの細胞の各々は、23の染色体を有する2つの他の細胞を産生する
- 1つの二倍体幹細胞は、4つの一倍体娘細胞を産生する
こちらの記事もご覧ください:更年期障害の発症を加速させる6つの要因
細胞はどのように染色体を失うのか?
一般的には「第2XまたはY染色体の喪失は受胎後に起こる」という理論があります。つまりこれは細胞分裂の失敗と定義されます。
Y染色体の欠如により女性の性的特性の発達が起こるため、元の細胞がXYであっても、染色体の損失により女性としての成長につながります。
ターナー症候群の臨床症状

小児期および成人期での成長の遅延
- 「スフィンクス」のような特徴の顔、扁平な鼻橋、低いの目、およびまぶたのわずかな奇形
- 通常よりも短くて幅の広い首
- 低いヘアライン
- 多数の色素沈着したほくろ
- 幅広い胸郭 乳児のような外見に伴う、離れた未発達の乳頭。
第二次性徴後の性的特製は非常に未成熟であり、性的幼児期が存在する - エストロゲンの不足により胸の発達はない。
- 陰毛はほとんどなく、丸みのある腰が特徴となる
- 骨の奇形は非常に一般的で、外向きの腕などがその一例となる
こちらの記事もご参考に:強い女性の6つの特徴
内部奇形
残念なことに、ターナー症候群を発症すると次のような体内の障害を併発する可能性があります。
- 大動脈縮径および他の弁奇形などの先天性心疾患が非常に一般的
- 多くの患者が腎臓の病気や問題を発症し、最も一般的には馬蹄鉄腎として発現する
- 性腺の発育異常:卵巣が十分に発達せず、代わりに生殖腺帯が存在する
- エストロゲン産生に必要なすべての刺激物は存在するものの、卵巣がないこために産生は不可能
- 無月経と不妊症:わずかな割合の患者のみが妊娠可能であり、ほとんどの患者が無月経で不妊なのがターナー症候群の最も一般的な特徴の1つとなる
ターナー症候群の診断

ターナー症候群を発症している女性のほとんどは明らかな疾患の特徴を持たず、軽度の症状しか現れないことが多いため、ターナー症候群は診断は非常に困難な場合が多くあります。
症例の約3分の1が新生児の時点で、ターナー症候群であると診断されます。
特定の身体的特徴の存在および心臓病の疑わしい症状などから判定できます。または他の3分の1の症例は、主に身長が低いために小児期に診断が確定します。
最後の3分の1は青年期に診断されます。 これらの症例では、性的乳児期と組み合わせた低身長は、患者が思春期に突入しても、身体の成長が平均的な状態へと到達しないときにはっきりします。
治療法
エストロゲンを使用するホルモン補充療法は、性的特性の発達に役立ちますが、不妊症を改善することはできません。 幼児期に診断された患者には、成長ホルモンを使って身長を正常化させることが推奨されています。
子供を持つことはできますか?
はい、ターナー症候群を発症している女性の中には子供を持つ可能性がある人もいます。 ターナー症候群の患者のほんの一部は、問題なく妊娠することできます。
ただし、一般的にはターナー症候群は不妊を伴っているケースが多く、海外では卵子の提供を受けて体外受精をする患者がいます。卵子はインビトロで精子と受精された後、患者に移植されます。ターナー症候群患者の妊娠は、正常な妊娠よりはるかに厳重なコントロールが必要とされています。
引用された全ての情報源は、品質、信頼性、時代性、および妥当性を確保するために、私たちのチームによって綿密に審査されました。この記事の参考文献は、学術的または科学的に正確で信頼性があると考えられています。
- Wyss D, DeLozier CD, Daniell J, Engel E. Structural anomalies of the X chromosome: personal observation and review of non-mosaic cases. Clin Genet. 2009; 21(2):145-59.
- Jacobs PA, Betts PR, Cockwell AE, Crolla JA, Mackenzie MJ, Robinson DO, et al. A cytogenetic and molecular reappraisal of a series of patients with Turner’s syndrome. Ann Hum Genet. 1999; 54(Pt 3):209-23.
- Lippe BM. Turner’s syndrome. In: Sperling M. Pediatric endocrinology. 3 ed. Philadelphia: Saunders; 2008.p. 387.
- Chagoyén Méndez, Esther María, Álvarez Montero, José Agustín, & Zúñiga Vaca, Carmen Isabel. (2017). Síndrome de Turner en una adolescente. MEDISAN, 21(6), 720-724. scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1029-30192017000600012&lng=es&tlng=es.